Share
First Model to Clearly Surpass AlphaFold 3 on Protein-Ligand Structure Prediction
Why is Structure Prediction a 'Holy Grail' Problem for Drug Discovery?
Creating a new medicine for patients is like creating a key for a complex biological lock. This “lock”—a target protein in the body—isn’t a rigid structure; it’s a flexible, dynamic puzzle that can change its shape when a molecule binds to it. Accurately predicting this 3D fit, a challenge known as protein-ligand structure prediction, would be transformative. It would allow scientists to move beyond slow, expensive trial-and-error and embrace rapid, rational design. By precisely modeling this interaction, researchers could digitally screen vast swaths of chemical space for potential drug candidates and engineer more effective medicines.
Today, we’re unveiling Pearl (Placing Every Atom in the Right Location) — a generative 3D foundation model for AI structure prediction that sets a new standard for biomolecular understanding. The model's full capabilities are described in our new technical report, co-authored with subject matter experts from NVIDIA.
Pearl Sets a New Standard for Structural Prediction
Since the 2024 Nobel Prize in Chemistry and the release of AlphaFold 3, the AI community has raced to replicate its powerful capabilities. It's a high bar to clear, and many attempts to date have yet to achieve true parity—often failing to reach sufficient accuracy to be useful for drug discovery, struggling to obey laws of physics such as predicting clashing atoms, or not effectively generalizing to novel drug targets.
Pearl is the first model to clearly surpass AlphaFold 3 on protein-ligand complex prediction, setting a new standard for this critical class of drug discovery tasks. Its advantages are even more evident in practical, real-world discovery workflows, where most existing models, including those evaluated here, are not robust enough for deployment. Below is a snapshot of Pearl’s performance (see our technical report for more details):
- New State-of-the-Art Cofolding Performance: Pearl significantly outperforms the other evaluated models for generating accurate and physically valid drug-protein poses on the public benchmarks, improving by 15% relative to AlphaFold 3 on Runs N’ Poses (Figure 1), and up to 40% relative improvement over AlphaFold 3 on PoseBusters (see our technical report).
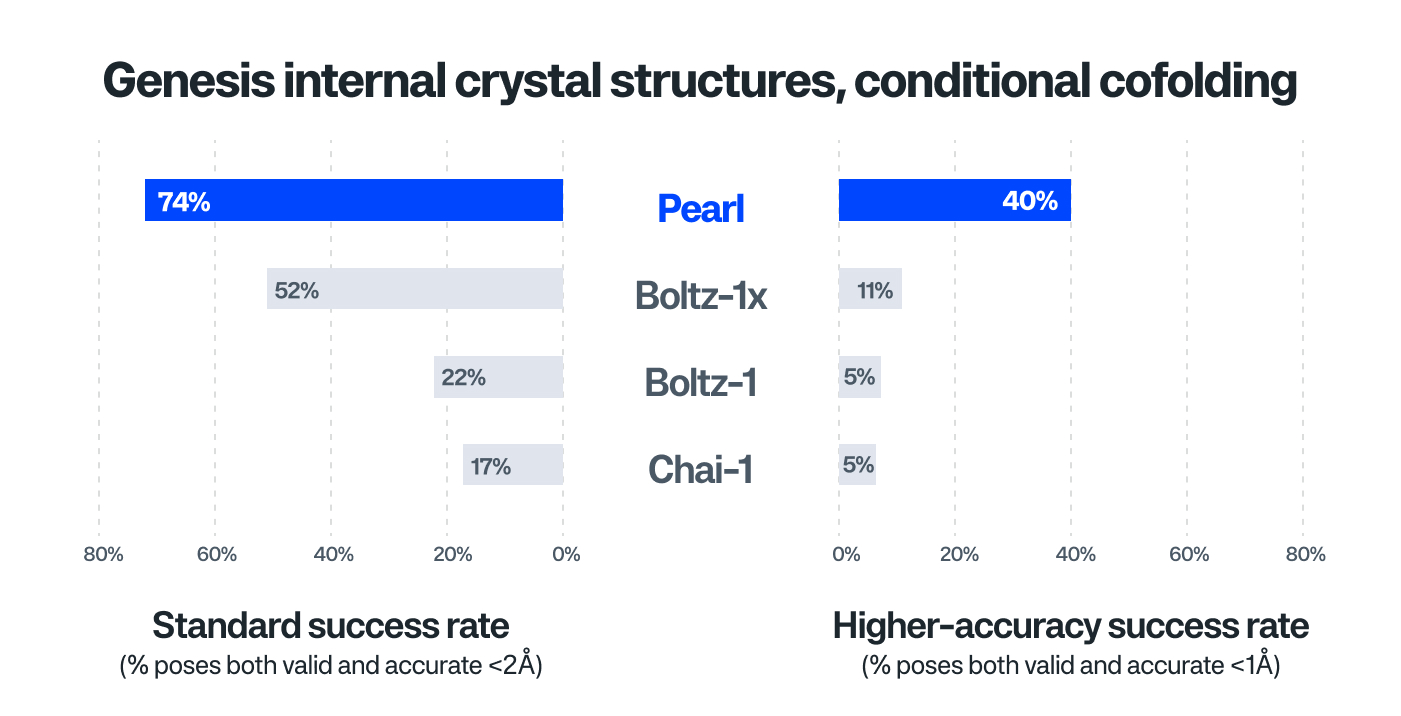
- Decisive Advantage when Deployed to Actual Pipeline Programs: Pearl uniquely maintains performance in real-world drug discovery (Genesis internal crystal structures), achieving over 40% relative improvement on standard success thresholds over the closest model (Figure 2).
- Unlocks Ultra-High Accuracy: Pearl distinguishes itself in achieving <1Å accuracy, not only for public benchmarks but also for internal crystal structures where its high-accuracy performance is several-fold more successful than the closest model (Figure 2). This capability is critical in enabling downstream utility such as using structures to predict potency.
Figure 1:
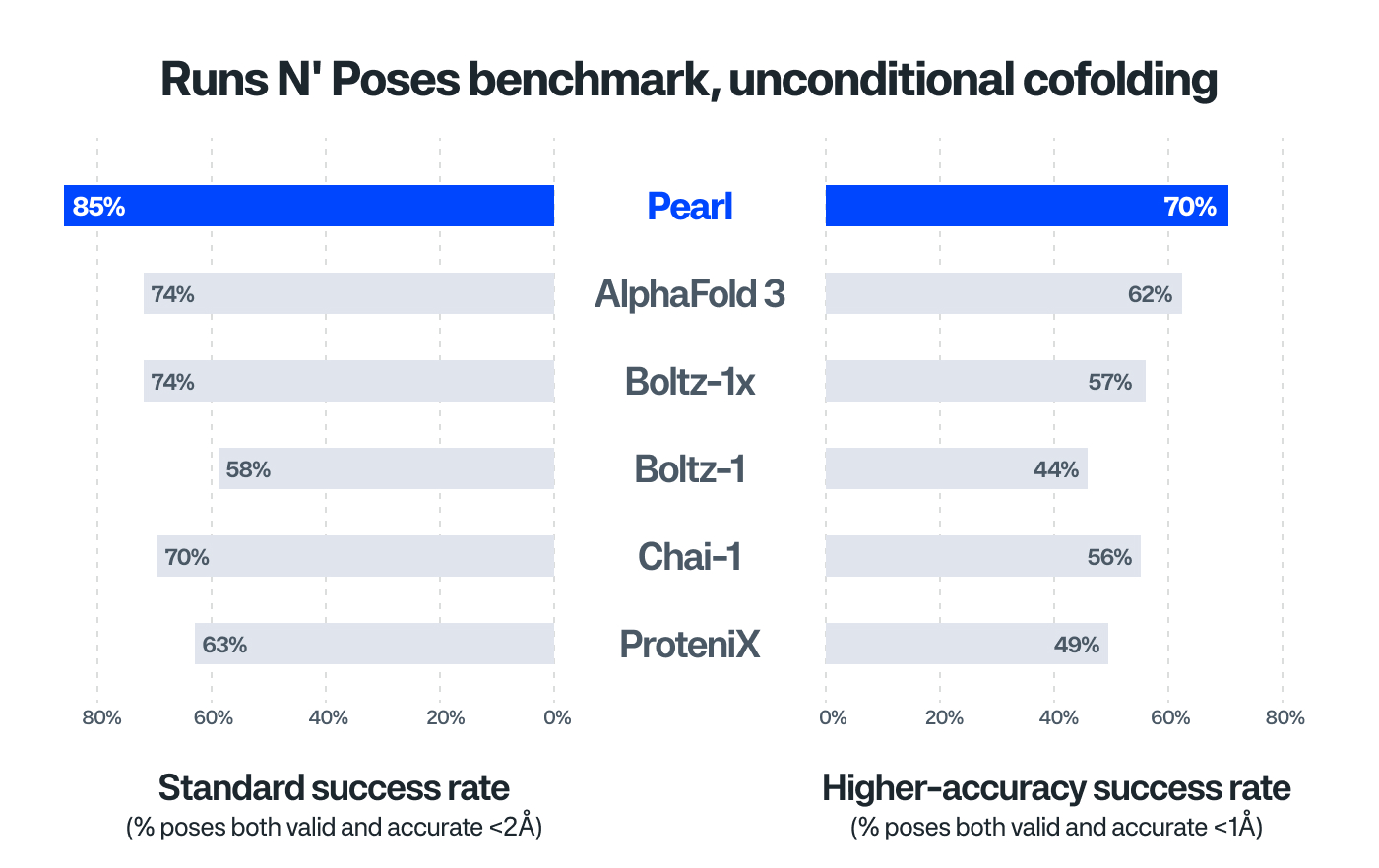
Figure 2:
Note: Models in Figures 1 & 2 were trained on public structures from the Protein Data Bank (PDB), but no PDB were used past a cutoff date of 2021-09-30, as is typical. A cutoff standard is important to perform fair comparisons of model architectures and to ensure training data is not leaked into benchmark test sets. However, our use of this cutoff is just for benchmarking; when we deploy Pearl in practice, we train a larger model on more PDB structures, as well as proprietary structures, to achieve maximum performance.
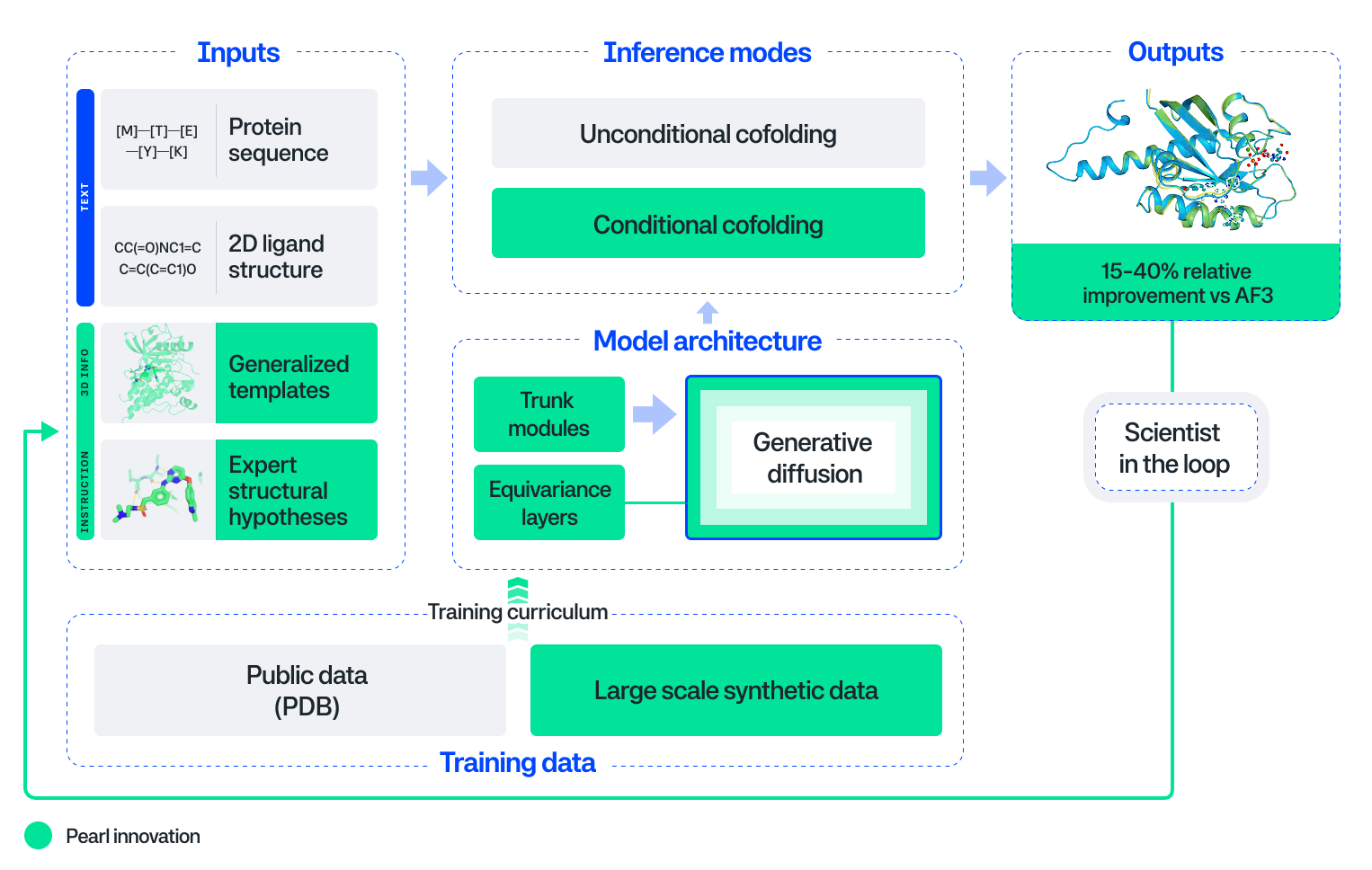
The Innovation Behind the Results
Pearl is a next generation 3D foundation model built to address the unique challenges of drug discovery. What truly sets Pearl apart is a suite of foundational machine learning innovations:
- Synthetic Data from Physics. Foundation models for language are trained on trillions of words. In stark contrast, the entire global repository of public protein-ligand structures—the data needed to train molecular AI—contains only a few hundred thousand examples. To overcome this, Pearl trains on a growing proprietary dataset of synthetic structures generated using physics, allowing it to learn from a chemical diversity that far exceeds what is possible from experiments alone, directly addressing a critical bottleneck.
- Evidence of Scaling Laws. We also show that we can continuously improve Pearl’s accuracy by adding more physics-generated synthetic complexes at training time. This is the first evidence of scaling laws for AI cofolding models using synthetic data, and it gives us a path to continue improving Pearl over time.
- Novel Architecture and Training for True Generalization. To ensure the model learns from this data instead of just memorizing it, Pearl uses a novel, proprietary architecture and training recipe. The architecture includes an SO(3)-equivariant diffusion head, which furnishes the model with geometric priors. In simple terms, this imparts 3D geometry directly into the model's DNA. It natively understands that a rotated molecule is still the same molecule, leading to more efficient training. The trunk leverages neural network modules that are more compute-efficient than those used in prior cofolding models, which unlocks greater model scale and performance. We also use NVIDIA cuEquivariance kernels (providing up to a 15% speedup for training and up to 80% speedup for inference in real-world scenarios).
- Unprecedented Scientist-in-the-Loop Control. A model is far more useful if it can be guided by expert intuition. Beyond basic prompting, Pearl provides scientists with a suite of advanced inference tools to put them "at-the-helm" of the discovery process. A key example is its generalized templating system enabling more rich in-context learning from prior structures of biomolecular complexes by directly leveraging information about non-protein components like co-factors and related ligands. Combined with inference-time steering techniques, this gives researchers the fine-grained control needed to test hypotheses, transforming Pearl from a black box into a dynamic scientific partner.
Pearl in Action: From Prediction to Discovery
Beyond its accuracy, Pearl is a practical tool designed for real-world drug discovery, offering two complementary modes that mirror R&D workflows:
- Unconditional Cofolding Mode: Pearl can predict a 3D structure using only the protein sequence and ligand 2D chemical structure. This is relevant for early-stage discovery before a binding pocket has been identified.
- Conditional Cofolding Mode: Scientists can also guide Pearl’s predictions by providing known information, like a specific binding pocket of interest or an experimental structure the user would like to use as a prompt. Pearl still generates a cofolding diffusion trajectory in conditional mode, but this 'in-context learning' for molecules allows researchers to test specific hypotheses on the fly and can significantly increase the accuracy.
Pearl is being deployed across both Genesis’ internal pipeline and in our partnership programs and is a cornerstone of the Genesis Exploration of Molecular Space (GEMS) platform. GEMS integrates structure prediction with molecule generation and property prediction, such as potency and key drug-like parameters. For drug hunters, this creates a powerful generative engine for discovery, enabling faster design cycles, more confident hypotheses, and the ability to unlock challenging protein targets that have not yet been drugged. Ultimately, Pearl can enable a fundamental shift from trial-and-error discovery to an era of rational design, with the goal of bringing next-generation medicines to patients faster than previously thought possible.